This is a very short tutorial discussing how to call variants using samtools and bcftools from BAM files in the context of a Whole Exome Sequencing (WES) experiment. All the following steps assume you are working in a Linux environment.
Software installation
samtools and bcftools may be downloaded from the following website: http://www.htslib.org/download/. Compiling and installing the software is easy and clearly explained on the website. Briefly, you can compile and install the softwares using the standard make and make install procedure. Don’t forget to add the installation directories to your $PATH variable to allow launching samtools and bcftools from any position.
cd samtools-1.x # and similarly for bcftools and htslib
sudo make
sudo make prefix=/where/to/install installexport PATH=/where/to/install/bin:$PATH # for sh or bash users
Variant Calling Workflow
The workflow is pretty straigthforward and can be summarized in 3 steps.
- Step 1. Generate pileups using the input BAM file and a fasta file including the sequence of the reference genome. The mpileup command (samtools) generates a BCF file that contains every locations in the genome
- Step 2. Remove the non-variant locations using bcftools
- Step 3. Prepare the VCF for querying by indexing it via tabix and/or perform some extra filtering.
The three-step procedure may be run as follows
samtools mpileup -ugf /data/mm10.fa -Q 20 /data/s2_sorted.bam | bcftools call -vmO z -o s2_variants.vcf.gz
tabix -p vcf s2_variants.vcf.gz
This will generate 2 files:
- a gzipped VCF file (.vcf.gz)
- a tabix index file for the previous file (.tbi)
Details
Let’s review the options and some details of the samtools and bcftools calls
- samtools mpileup
- -u, –uncompressed: generate uncompressed VCF/BCF output
- -g, –BCF: generate genotype likelihoods in BCF format. Alternatively, samtools may be run using the option -v, –VCF to generate genotype likelihoods in VCF format. However the resulting output differs significantly from our final VCF file (indeed, no filtering for the non-variant sites is performed at this step)
- -Q, –min-BQ: skip positions with base quality lower than the number provided as argument
- bcftools call
- -v ()-v, –variants-only output variant sites only
- -m mutliallelic (alternative model for multiallelic and rare-variant calling designed to overcome known limitations in -c calling)
- -O type of output: Output compressed BCF (b), uncompressed BCF (u), compressed VCF (z), uncompressed VCF (v)
- -o name of the output file
- At this stage, you can selectivey remove indels or point variants by including respectively one of the following flags: –skip-variants snps | indels or -V snps | -V indels
The VCF file generated following this tutorial will look somewhat like this
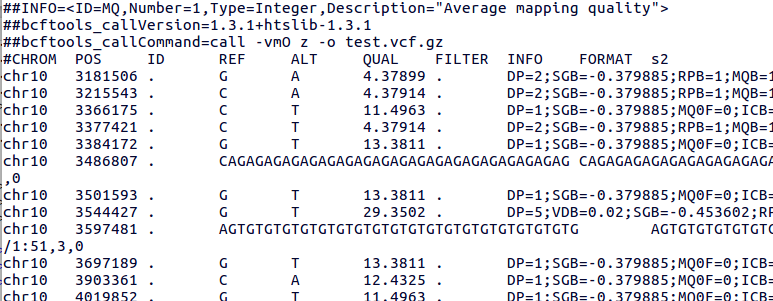
Notes
When running samtools on BAM files generated by someone else, make sure that the sequence names in the fasta files are the same as those in the BAM files. Positions on unmatched sequences (unrecognized chromosome names) will be filtered out.
References and more information can be found here: