Project Goal and Data Understanding
The goal of this project is to predict patients response to the anti-HIV treatment based on a very limited number of measured parameters. The dataset was retrieved from Kaggle (https://www.kaggle.com/c/hivprogression).
- Dataset includes information about a patient’s viral load, CD4 count and the nucleotide sequences of their Reverse Transcriptase (RT) and their Protease (PR) HIV genes.
- In this project, false positives and a false negatives are weighted equally.
- The goal is to manage to extract useful information from the DNA sequences. These features may be easy to process using specific software, but may be difficult to annotate in a large corpus of sequences like this one. Datasets were downloaded from: https://www.kaggle.com/c/hivprogression/data
Data preparation and Data Cleaning (summary)
Viral load and Log(CD4+ count) data were used as is. On the contrary, PR and TR nucleotide sequences were analyzed in order to extract biologically meaningful information. New features were extracted by analyzing the dataset with R.
PR
Most of the samples have a 297 bp long PR sequence. Only few sequences bearing deletions were found. Some sequences were missing. A consensus sequence was defined starting from the sequences found in the training set. Frequently mutated regions were identified and analyzed. Extracted features included:
- distance from consensus
- nucleotide present at hyper-variable sites
- distance of hyper-variable regions from consensus
- %GC of hyper-variable regions
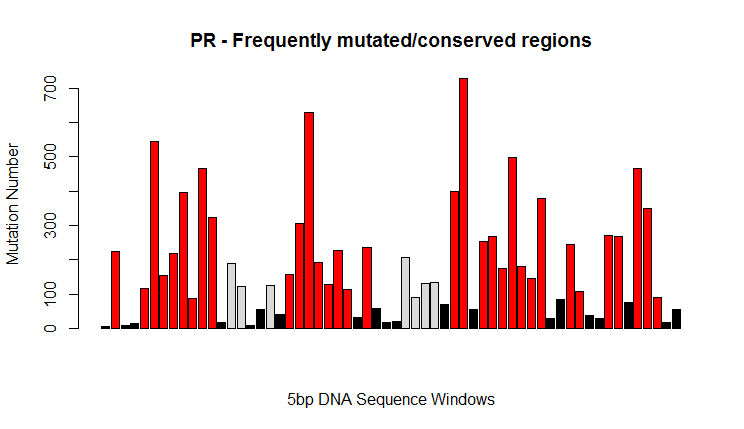
Features based on missing PR sequences were imputed using the medians (for categorical variables, the most frequent value was used) of the rest of the cases in the train subset.
RT
RT sequence is extremely variable. Nucleotide length of RT sequences spans a very wide range (600 bp to 1600 bp). Because of this, a consensus is almost impossible to define. In the end, the following features were extracted for each PR sequence:
- sequence length in bp
- %GC of each sequence
- % of mismatches/non assigned bases in the sequence (% of non-A,T,C,G letters in the sequence)
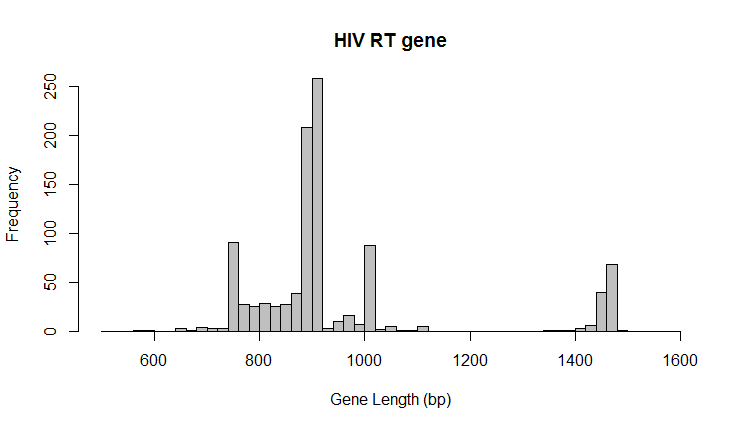 Modeling and deployment (summary)
Modeling and deployment (summary)
A Neural Network Model was built using Statistica13 software (Dell). For building the model, the following steps were preformed:
- The TRAIN dataset was sampled in order to obtain a subset balanced for the target variable (response)
- Features were analyzed by applying the Feature Selection Tool
- Performances of three different modeling algorithms (neural network, SVM and boosted trees) were compared: the Neural Network algorithm gave the best results
- A Classification Neural Network Model was built and deployed on the TEST dataset (retrieved from Kaggle)
Interestingly, the inclusion of features derived from the analysis of PR and RT nucleotide sequences greatly improved the performance of the Neural Network Model
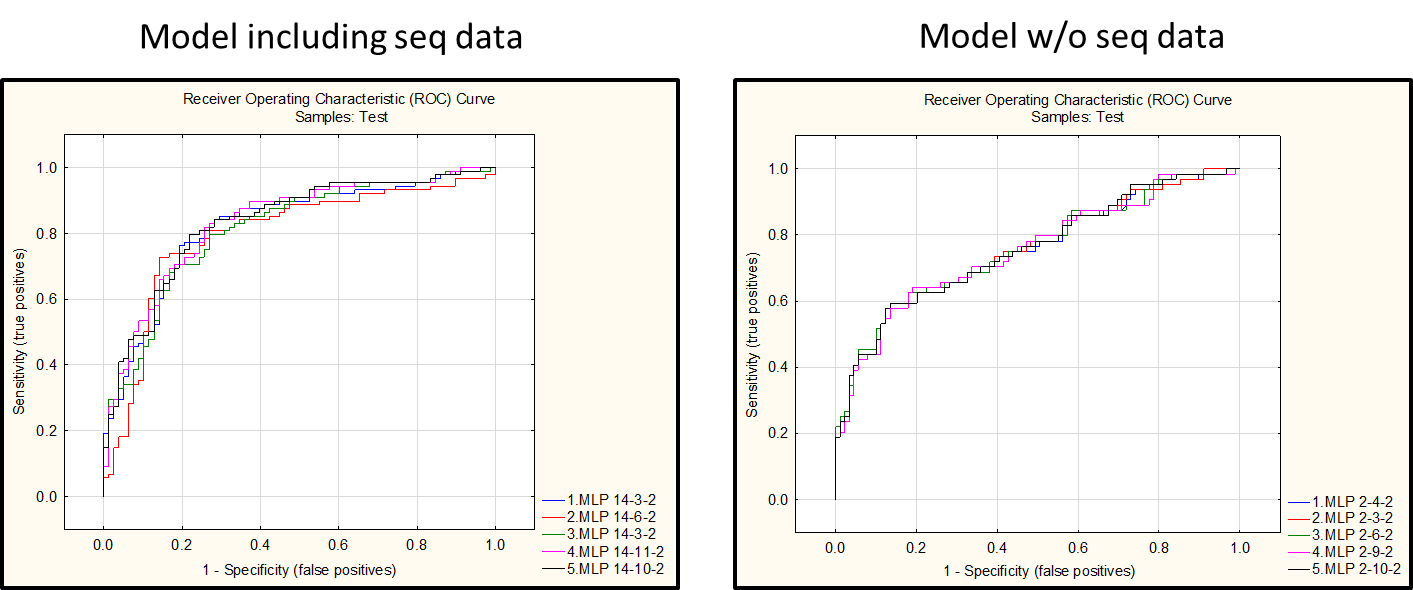
Conclusions
- A model for predicting response to antiviral treatment of HIV patients was generated. This model is based on 2 clinical parameters measured at the time of the initial treatment (initial viral load and CD4+ cell counts in patient’s blood) as well as on the nucleotide sequences of 2 viral genes: PR and RT.
- The initial viral load is the major predictor
- Several features extracted by annotating HIV PR and TR nucleotide sequences were employed for building the model. For example, high sequence variability in specific regions of the PR gene sequence (such as r7: 240-250bp) correlates with a poor response (it could be interesting to further analyze the underlying biological mechanisms, as there could be some potential for drug development).
- The model was built using a neural network algorithm and has an error rate of about 25% in the test set.
Supporting documents
A detailed step-by-step description of the project is available here: HIV tutorial – dfantini_2016