Homolog genes are genes descending from a common ancestral DNA sequence. Homolog genes in different species (i.e., genes that evolved from a common ancestral gene by speciation) are referred to as ortholog genes and usually retain a very similar function in the course of evolution. On the contrary, paralog genes are homolog genes present in the same genome (obtained by gene duplication). Paralog genes may have a similar function but also evolve distinctive features (http://medblog.stanford.edu/lane-faq/archives/2006/05/what_are_orthol.html).
Sometimes, we may want to compare homolog genes. For this, given a gene symbol, we may want to retrieve information about:
- which are its paralog genes (in the same genome)?
- which are its ortholog genes (in a different genome)?
We can use biomaRt to retrieve these information. BiomaRt (https://bioconductor.org/packages/release/bioc/vignettes/biomaRt/inst/doc/biomaRt.html) provides a powerful yet simple-to-use interface to a growing collection of databases that include Ensembl, Uniprot and HapMap. BiomaRt allows to retrieve a wide panel of different types of information. Here, only some basic examples and the retrieval of homolog genes will be covered. For more examples, you may look up the following web page: http://pevsnerlab.kennedykrieger.org/wiley/3e/chapter8/WebDocument_8-01_biomaRt.Rmd.
Let’s start by loading biomaRt and selecting the ensembl database. Next, we will select the human (hsapiens_gene_ensembl) and the murine (mmusculus_gene_ensembl) datasets.
library(biomaRt)
ensembl <- useMart(“ensembl”)
ensembl.human <- useMart(“ensembl”, dataset = “hsapiens_gene_ensembl”)
ensembl.mouse <- useMart(“ensembl”, dataset = “mmusculus_gene_ensembl”)
A standard biomaRt query will be structured as follows. Briefly, we will use the getBM function to query the biomaRt dataset of choice. The query requires the following arguments to be passed in:
- attributes: identifier(s) of the field(s) we want to retrieve
- filters: field identifier of the supplied values used for the query (correspond to keytype, in a select OrganismDbi::select() query)
- values: query values (correspond to keys, in a select OrganismDbi::select() query)
- mart: dataset of choice
getBM(attributes = c(‘hgnc_symbol’, ‘chromosome_name’, ‘start_position’, ‘end_position’),
filters = ‘hgnc_symbol’,
values = c(“TP53”, “XPC”, “DDB1”, “APEX1”),
mart = human.ensembl)
In order to retrieve paralog genes from the same genome, we should search for fields (attributes) that include “paralog” information.
BM.attributes = listAttributes(human.ensembl)
grep(“paralog”, tolower(BM.attributes[,1]), value = TRUE)
#
# we may use the following field: hsapiens_paralog_associated_gene_name
getBM(attributes = c(‘hsapiens_paralog_associated_gene_name’, ‘hsapiens_paralog_chromosome’),
filters = ‘hgnc_symbol’,
values = c(“E2F1”),
mart = human.ensembl)#
# hsapiens_paralog_associated_gene_name hsapiens_paralog_chromosome
# 1 E2F2 1
# 2 E2F3 6
# 3 E2F6 2
# 4 E2F4 16
# 5 E2F5 8
Likewise, in order to retrieve ortholog genes from a different genome we can do as follows.
getBM(attributes = c(‘mmusculus_homolog_associated_gene_name’, ‘mmusculus_homolog_chromosome’),
filters = ‘hgnc_symbol’,
values = c(“E2F3”),
mart = human.ensembl)#
# mmusculus_homolog_associated_gene_name mmusculus_homolog_chromosome
# 1 E2f3 13
If we want, we can join data from different datasets and perform a JOIN query. Please, note that this type of query will take much longer than a one-dataset (single table) query. JOIN queries are performed by specifying an attributesL and martL parameters.
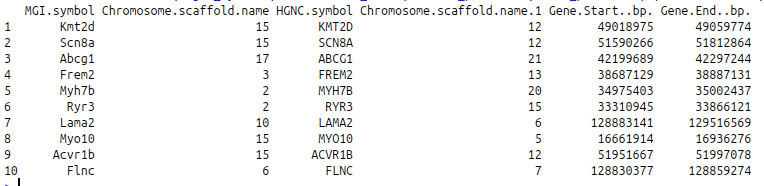
my.mouse.genes <- c(“Frem2”, “Kmt2d”, “Scn8a”, “Abcg1”, “Acvr1b”, “Flnc”, “Lama2”, “Myh7b”, “Myo10”, “Ryr3”)
getLDS(attributes = c(“mgi_symbol”, “chromosome_name”),
filters = “mgi_symbol”, values = my.mouse.genes, mart = mouse.ensembl,
attributesL = c(“hgnc_symbol”, “chromosome_name”, “start_position”, “end_position”) , martL = human.ensembl)
 Success!
Success!